Veuillez indiquer si vous êtes acheteur ou fournisseur pour trouver des ressources et des articles spécifiquement adaptés à vos besoins.
Comprendre les informations relatives à la biocompatibilité sur une page produit
Explication de l'étiquetage des dispositifs médicaux, des classes de risque et des procédures d'approbation
Lors de la sélection de biomatériaux ou de dispositifs médicaux, il est essentiel de comprendre les informations sur la biocompatibilité fournies sur la page produit. Ces données permettent d’évaluer si un produit répond aux exigences de sécurité, réglementaires et fonctionnelles pour des applications médicales. Voici comment interpréter les sections clés relatives à la biocompatibilité sur une page produit.
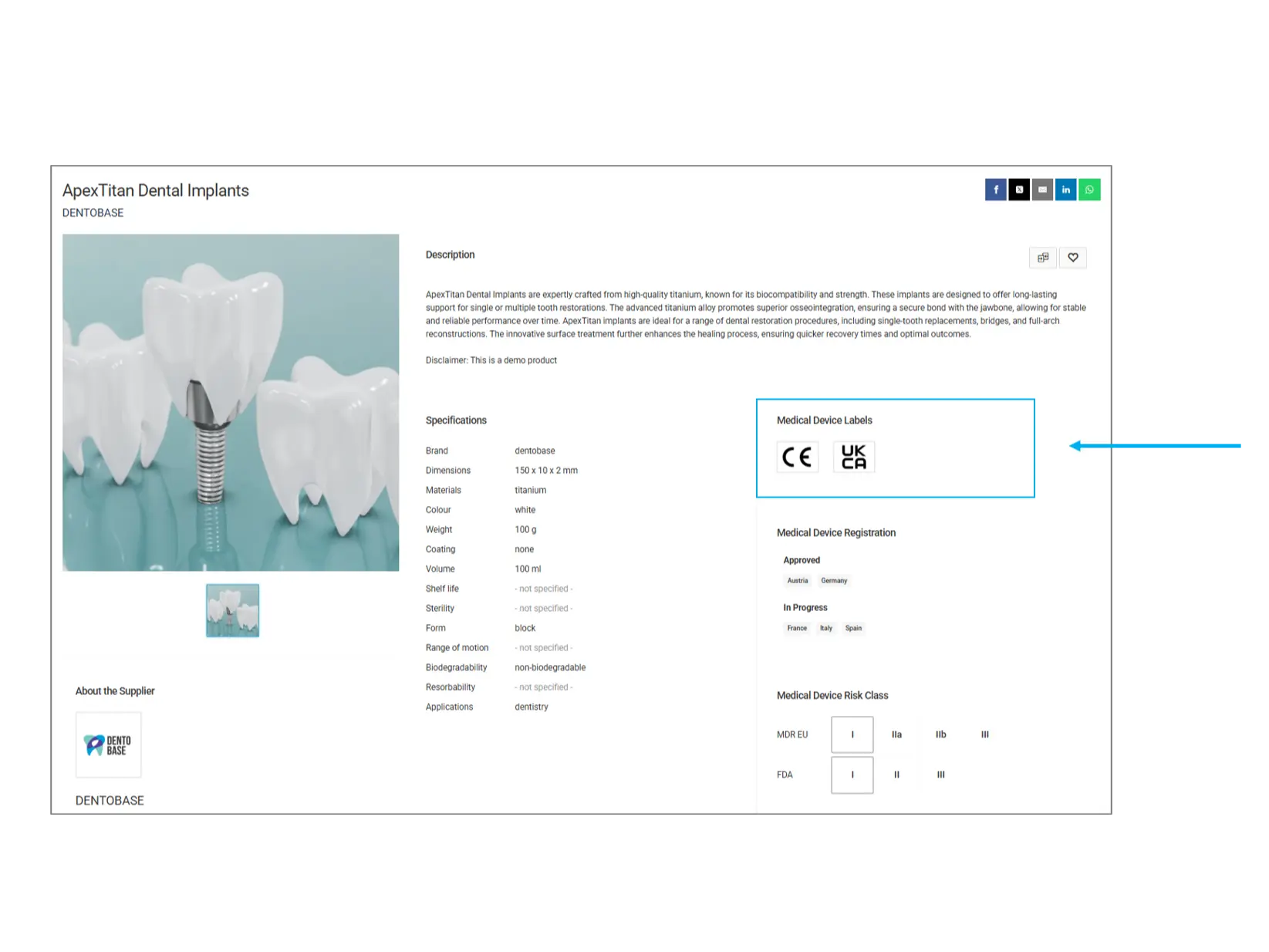
Étiquetage des dispositifs médicaux
Les étiquettes des dispositifs médicaux indiquent les normes réglementaires et les certifications respectées par un produit. Ces labels garantissent que le produit satisfait aux exigences de sécurité et de qualité requises.
CE (Conformité Européenne) : Indique que le produit est conforme aux normes de sécurité, de santé et de protection de l’environnement de l’UE.
UKCA (United Kingdom Conformity Assessed) : Certification pour les produits vendus au Royaume-Uni, garantissant la conformité aux réglementations britanniques.
Autres labels: Des certifications supplémentaires telles que la FDA, l’ISO ou d’autres approbations spécifiques à chaque pays peuvent être présentes.
À vérifier: Assurez-vous que le produit dispose des certifications requises selon les exigences réglementaires de votre pays.
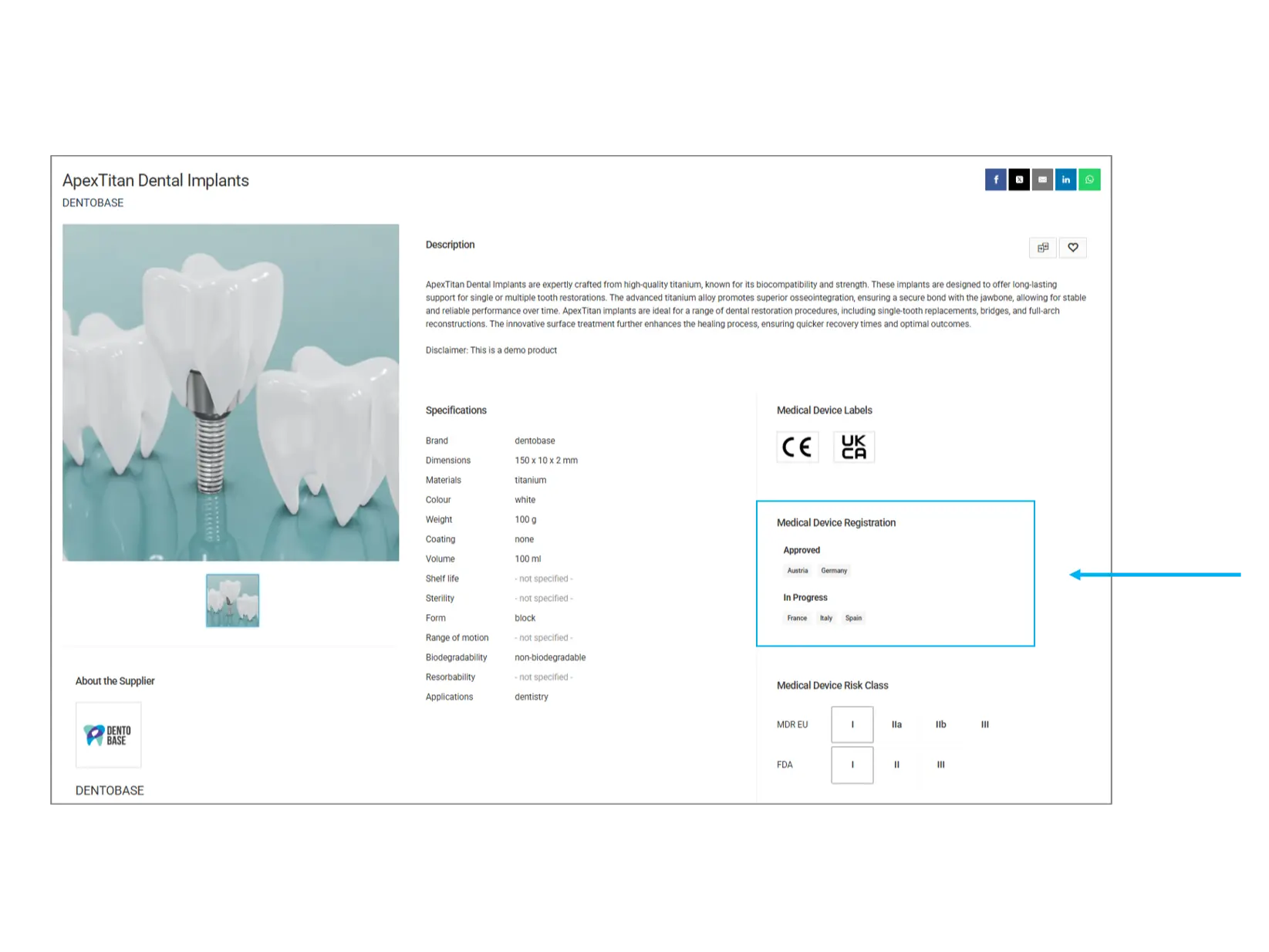
Enregistrement du dispositif médical
Cette section fournit des informations sur le statut d’approbation réglementaire du produit dans différents pays.
Pays approuvés: Le produit a été officiellement enregistré et peut être commercialisé dans ces régions.
En cours d’approbation: Le produit est en phase d’évaluation réglementaire et n’est pas encore totalement approuvé dans les pays indiqués.
À vérifier: Si vous avez besoin d’un produit pour un marché spécifique, assurez-vous qu’il est approuvé dans votre pays avant de l’acheter.
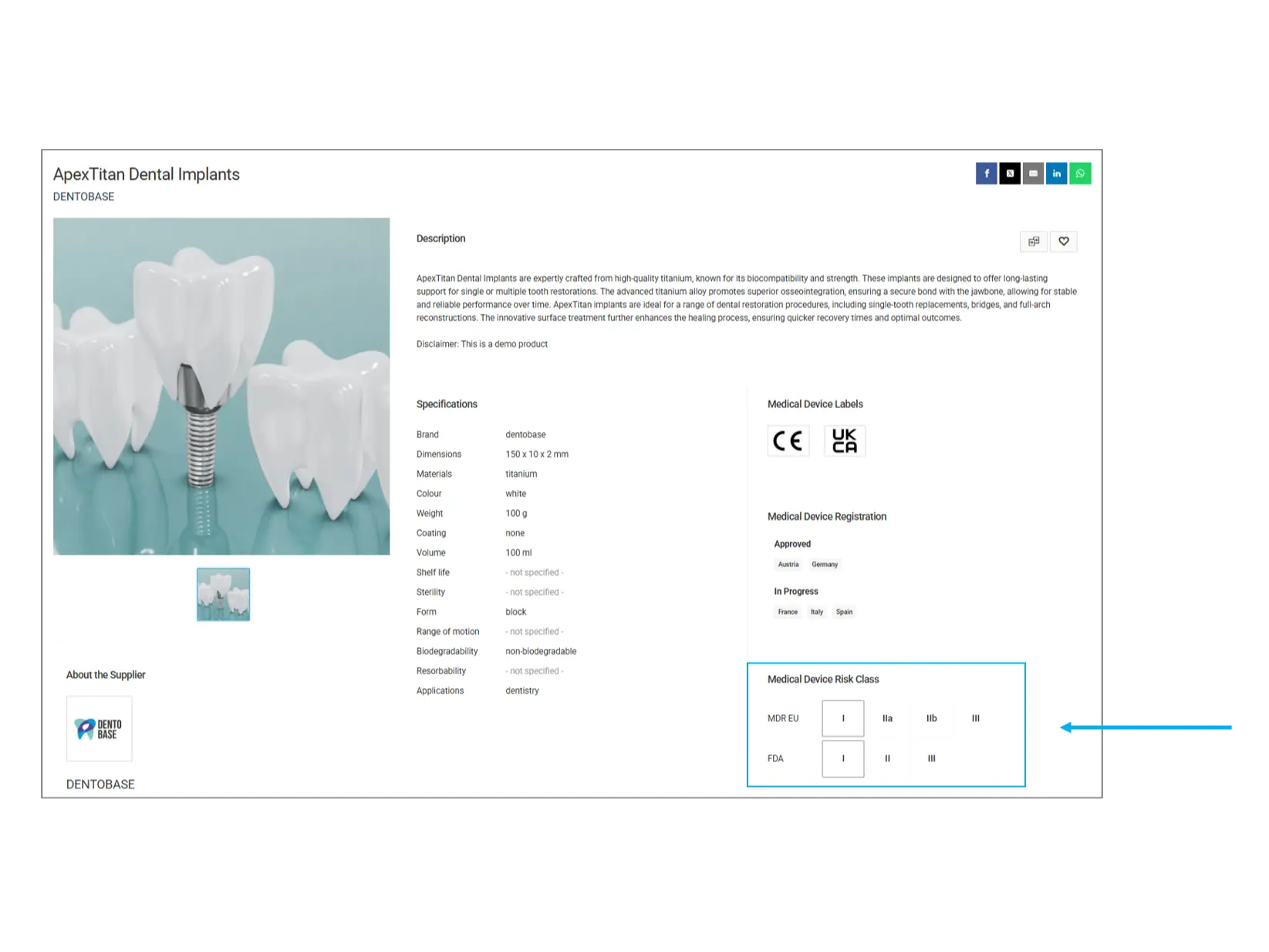
Classification des risques des dispositifs médicaux
Les dispositifs médicaux sont classés en différentes catégories de risque en fonction de leur impact potentiel sur la santé humaine. Cette classification permet de comprendre le niveau de réglementation et les risques potentiels associés au produit.
MDR UE (Règlement sur les dispositifs médicaux - Union européenne):
Classe I: Faible risque (ex. pansements, instruments dentaires)
Classe IIa: Risque modéré (ex. appareils auditifs, imagerie diagnostique)
Classe IIb: Risque élevé (ex. pompes à perfusion, ventilateurs)
Classe III: Risque très élevé (ex. stimulateurs cardiaques, valves cardiaques artificielles)
FDA (Food and Drug Administration - États-Unis):
Classe I: Contrôles généraux (ex. abaisse-langue, gants chirurgicaux)
Classe II: Contrôles spécifiques (ex. tensiomètres, pompes à perfusion)
Classe III: Nécessite une approbation préalable (ex. défibrillateurs implantables, valves cardiaques)
À vérifier: Les dispositifs de classe supérieure sont soumis à des réglementations et à des tests plus stricts. Si votre application nécessite un produit hautement réglementé, assurez-vous qu’il répond à la classification requise.
Is this article helpful?