Seleziona se sei un acquirente o un fornitore per trovare risorse e articoli specificamente pensati per le tue esigenze.
Comprendere le informazioni sulla biocompatibilità nella pagina di un prodotto
Spiegazione dell'etichettatura dei dispositivi medici, delle classi di rischio e delle procedure di approvazione.
Quando si selezionano biomateriali o dispositivi medici, è fondamentale comprendere le informazioni sulla biocompatibilità fornite nella pagina del prodotto. Questi dati aiutano a valutare se un prodotto soddisfa i requisiti di sicurezza, normativi e funzionali per applicazioni mediche. Di seguito viene spiegato come interpretare le sezioni chiave relative alla biocompatibilità in una pagina di prodotto.
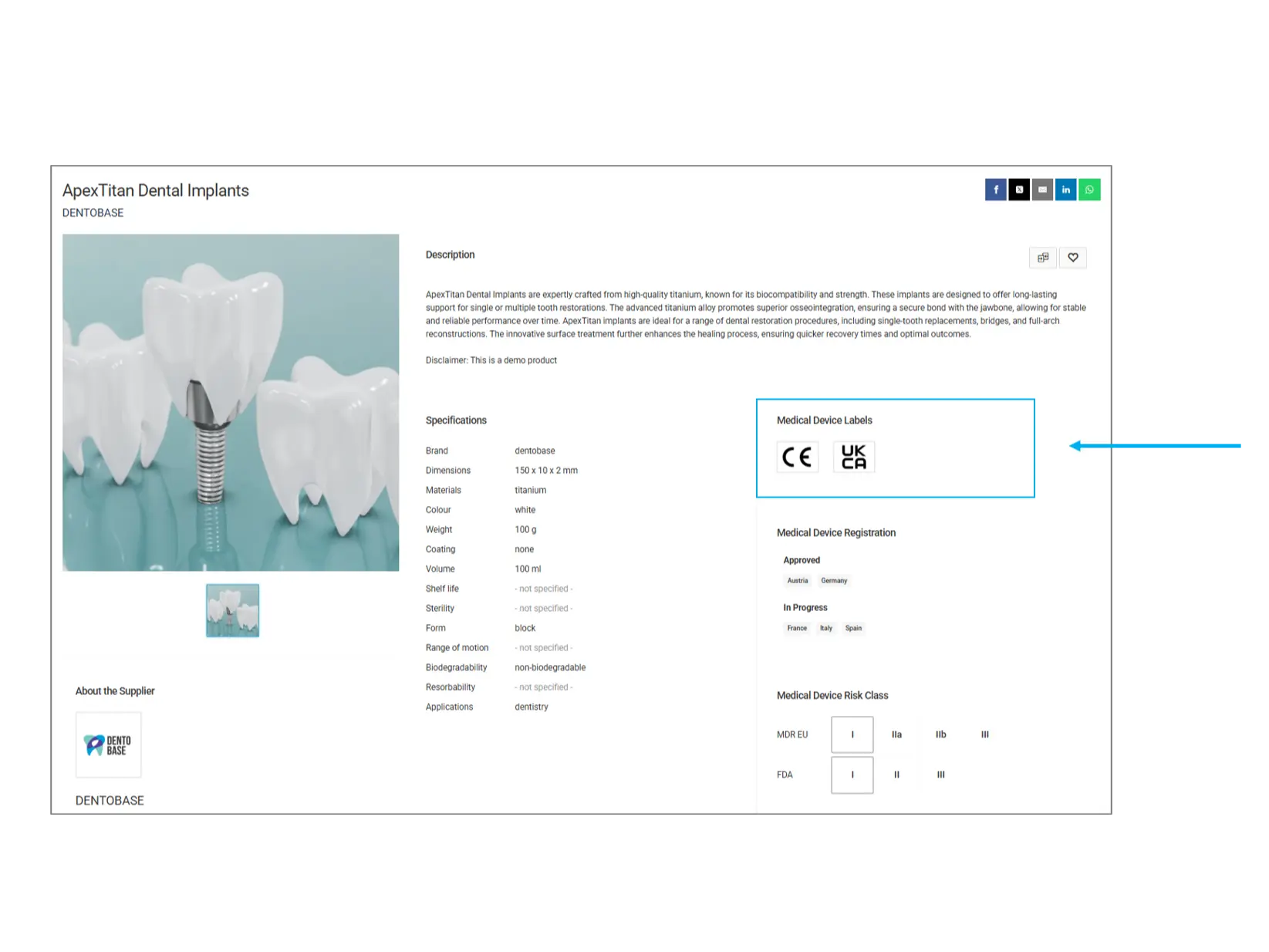
Etichette dei dispositivi medici
Le etichette dei dispositivi medici indicano gli standard normativi e le certificazioni rispettate da un prodotto. Queste etichette garantiscono che il prodotto soddisfi i requisiti di sicurezza e qualità richiesti.
CE (Conformité Européenne): Indica che il prodotto è conforme agli standard di sicurezza, salute e protezione ambientale dell'UE.
UKCA (United Kingdom Conformity Assessed): Certificazione per prodotti venduti nel Regno Unito, che ne garantisce la conformità alle normative britanniche.
Altre etichette: Possono essere presenti certificazioni aggiuntive come FDA, ISO o altre approvazioni specifiche di ciascun paese.
Cosa controllare? Assicurati che il prodotto abbia le certificazioni richieste in base ai requisiti normativi del tuo paese.
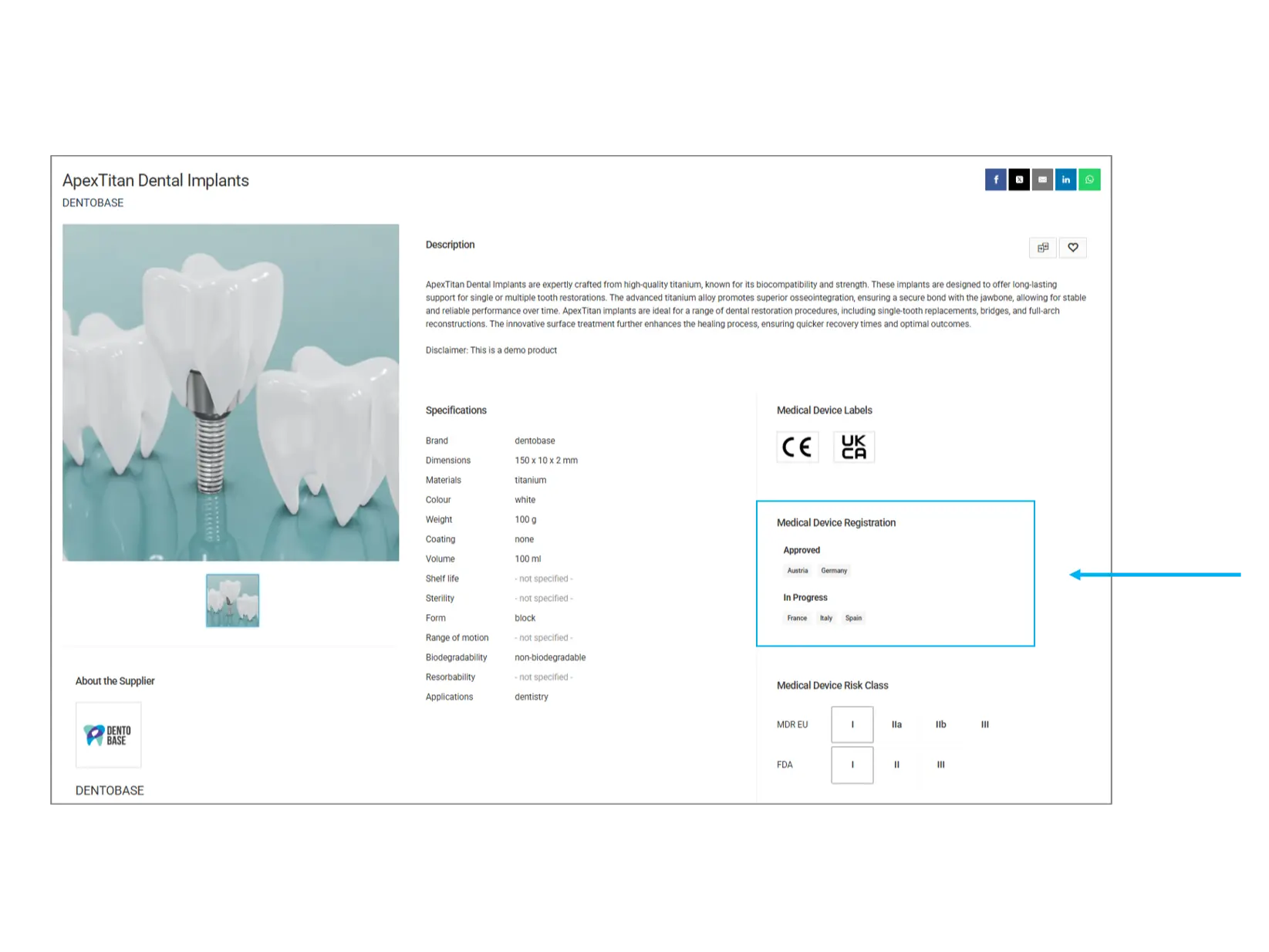
Registrazione del dispositivo medico
Questa sezione fornisce informazioni sullo stato di approvazione normativa del prodotto in diversi paesi.
Paesi approvati: Il prodotto è stato ufficialmente registrato e può essere commercializzato in queste regioni.
In fase di approvazione: Il prodotto è in fase di valutazione normativa e non è ancora completamente approvato nei paesi elencati.
Cosa controllare? Se hai bisogno di un prodotto per un mercato specifico, verifica che sia approvato nel tuo paese prima dell'acquisto.
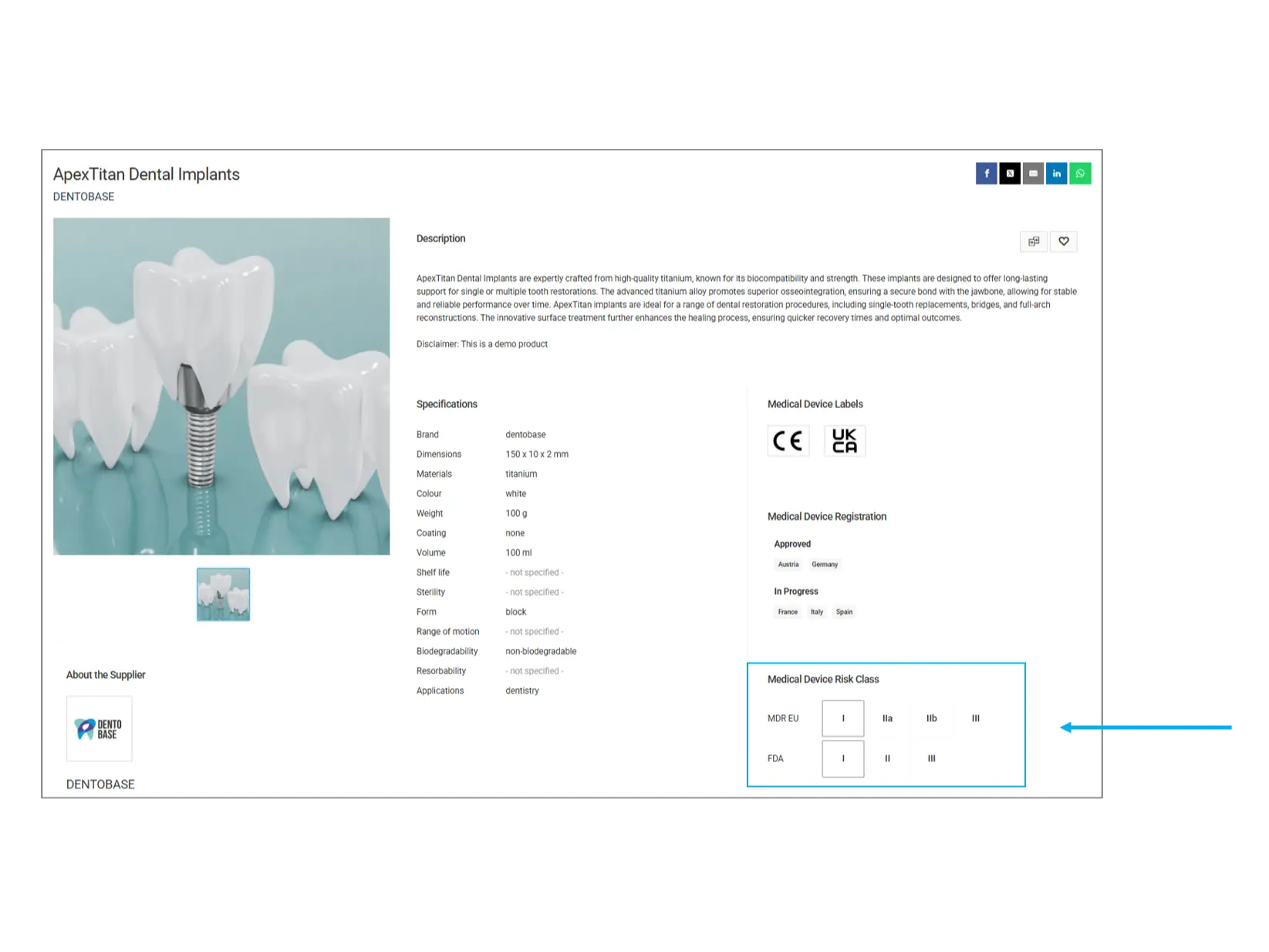
Classificazione del rischio dei dispositivi medici
I dispositivi medici sono classificati in diverse categorie di rischio in base al loro potenziale impatto sulla salute umana. Questa classificazione aiuta a comprendere il livello di regolamentazione e i possibili rischi associati al prodotto.
MDR UE (Regolamento Dispositivi Medici - Unione Europea):
Classe I: Rischio basso (es. bende, strumenti dentali)
Classe IIa: Rischio medio (es. apparecchi acustici, imaging diagnostico)
Classe IIb: Rischio elevato (es. pompe per infusione, ventilatori)
Classe III: Rischio molto alto (es. pacemaker, valvole cardiache artificiali)
FDA (Food and Drug Administration - Stati Uniti):
Classe I: Controlli generali (es. abbassalingua, guanti chirurgici)
Classe II: Controlli speciali (es. sfigmomanometri, pompe per infusione)
Classe III: Richiede approvazione preventiva (es. defibrillatori impiantabili, valvole cardiache)
Cosa controllare? I dispositivi di classe superiore sono soggetti a normative e test più rigorosi. Se la tua applicazione richiede un prodotto altamente regolamentato, assicurati che soddisfi la classificazione necessaria.
Is this article helpful?