Bitte wählen Sie aus, ob Sie Käufer oder Anbieter sind, um Ressourcen und Artikel zu finden, die speziell auf Ihre Bedürfnisse zugeschnitten sind.
Biokompatibilitätsbezogene Informationen auf einer Produktseite verstehen
Erläuterung der Kennzeichnung von Medizinprodukten, Risikoklassen und Zulassungsverfahren
Bei der Auswahl von Biomaterialien oder Medizinprodukten ist es wichtig, die auf der Produktseite angegebenen biokompatibilitätsbezogenen Informationen zu verstehen. Diese Daten helfen dabei, zu bewerten, ob ein Produkt den Sicherheits-, Regulierungs- und Funktionsanforderungen für medizinische Anwendungen entspricht. Nachfolgend finden Sie eine Übersicht über die wichtigsten biokompatibilitätsbezogenen Abschnitte einer Produktseite und deren Bedeutung.
Medizinische Gerätezertifizierungen (Labels)
Medizinische Gerätezertifizierungen zeigen an, welchen regulatorischen Standards ein Produkt entspricht. Diese Labels stellen sicher, dass das Produkt die erforderlichen Sicherheits- und Qualitätsrichtlinien erfüllt.
CE (Conformité Européenne): Zeigt an, dass das Produkt den EU-Sicherheits-, Gesundheits- und Umweltschutzstandards entspricht.
UKCA (United Kingdom Conformity Assessed): Eine Zertifizierung für Produkte, die in Großbritannien verkauft werden, um die Einhaltung der britischen Vorschriften sicherzustellen.
Weitere Labels: Zusätzliche Zertifizierungen wie FDA, ISO oder andere länderspezifische Zulassungen können vorhanden sein.
Worauf sollten Sie achten? Stellen Sie sicher, dass das Produkt über die erforderlichen Zertifizierungen für Ihr Land oder Ihre regulatorischen Anforderungen verfügt.
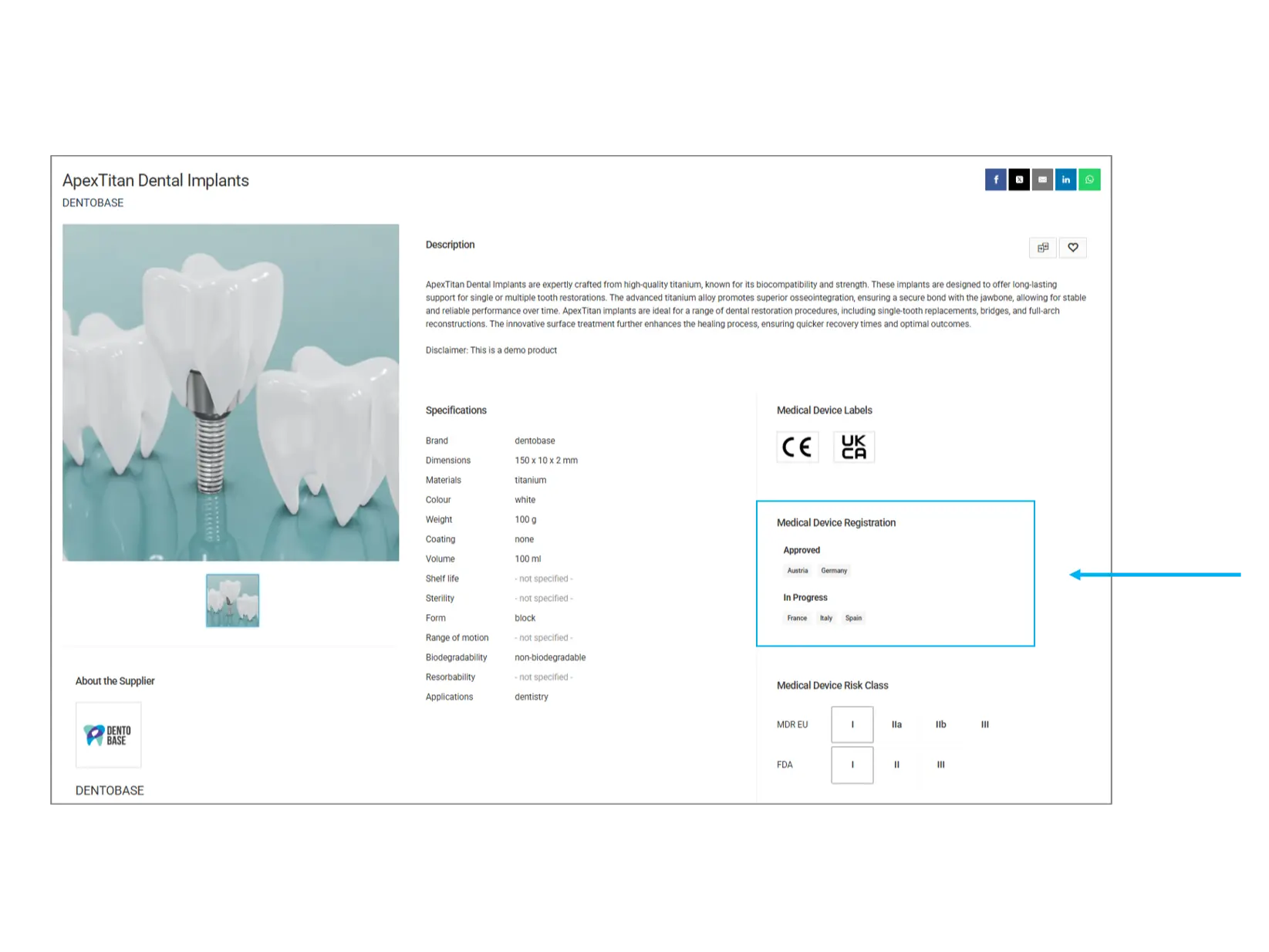
Medizinische Gerätezulassung
Dieser Abschnitt gibt an, in welchen Ländern das Produkt bereits behördlich zugelassen ist oder sich noch im Zulassungsverfahren befindet.
Genehmigte Länder: Das Produkt wurde offiziell registriert und kann in diesen Regionen verkauft werden.
In Bearbeitung: Das Produkt befindet sich im Prüfverfahren und ist in den aufgeführten Ländern noch nicht vollständig zugelassen.
Worauf sollten Sie achten? Falls Sie das Produkt für einen bestimmten Markt benötigen, prüfen Sie, ob es in Ihrem Land zugelassen ist, bevor Sie es kaufen.
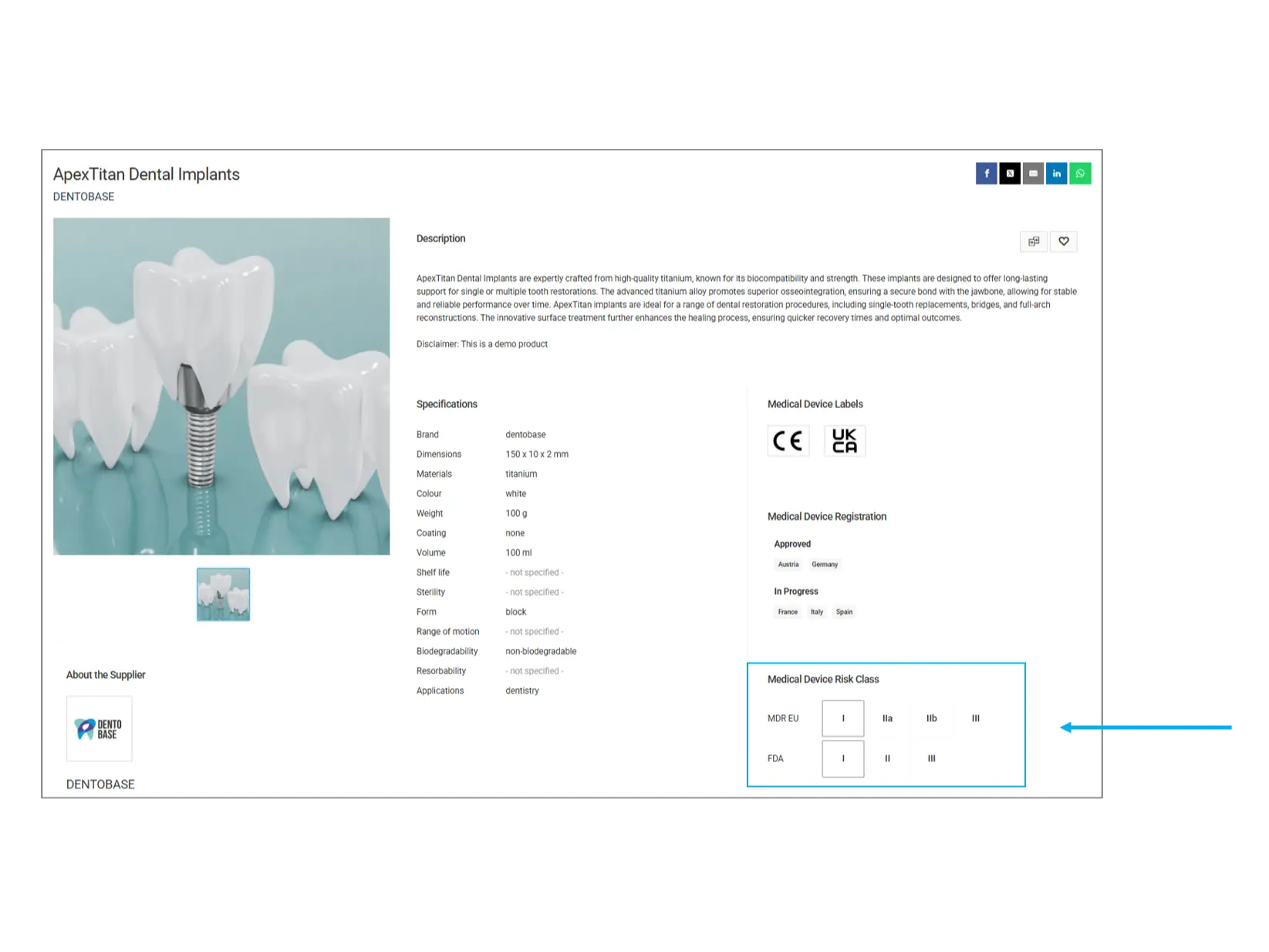
Risikoklassifizierung von Medizinprodukten
Medizinprodukte werden anhand ihres Risikopotenzials in verschiedene Kategorien eingeteilt. Diese Klassifizierung hilft dabei, die regulatorischen Anforderungen und potenziellen Risiken eines Produkts zu verstehen.
MDR EU (Medical Device Regulation - Europäische Union):
Klasse I: Geringes Risiko (z. B. Verbandsmaterial, Dentalinstrumente)
Klasse IIa: Mittleres Risiko (z. B. Hörgeräte, diagnostische Bildgebung)
Klasse IIb: Höheres Risiko (z. B. Infusionspumpen, Beatmungsgeräte)
Klasse III: Hohes Risiko (z. B. Herzschrittmacher, künstliche Herzklappen)
FDA (U.S. Food and Drug Administration):
Klasse I: Allgemeine Vorschriften (z. B. Holzspatel, OP-Handschuhe)
Klasse II: Besondere Vorschriften (z. B. Blutdruckmessgeräte, Infusionspumpen)
Klasse III: Erfordert eine behördliche Zulassung (z. B. implantierbare Defibrillatoren, künstliche Herzklappen)
Worauf sollten Sie achten? Höher klassifizierte Geräte unterliegen strengeren Vorschriften und Tests. Falls Ihr Anwendungsbereich eine hohe Regulierungsstufe erfordert, stellen Sie sicher, dass das Produkt die notwendige Klassifizierung erfüllt.
Is this article helpful?