Please select whether you are a buyer or supplier to find resources and articles specifically tailored to your needs.
Understanding biocompatibility information on a product page
Explanation of medical device labelling, risk classes and approval procedures
When selecting biomaterials or medical devices, understanding the biocompatibility information provided on the product page is essential. This data helps assess whether a product meets safety, regulatory, and functional requirements for medical applications. Below is a breakdown of the key biocompatibility-related sections found on a product page and how to interpret them.
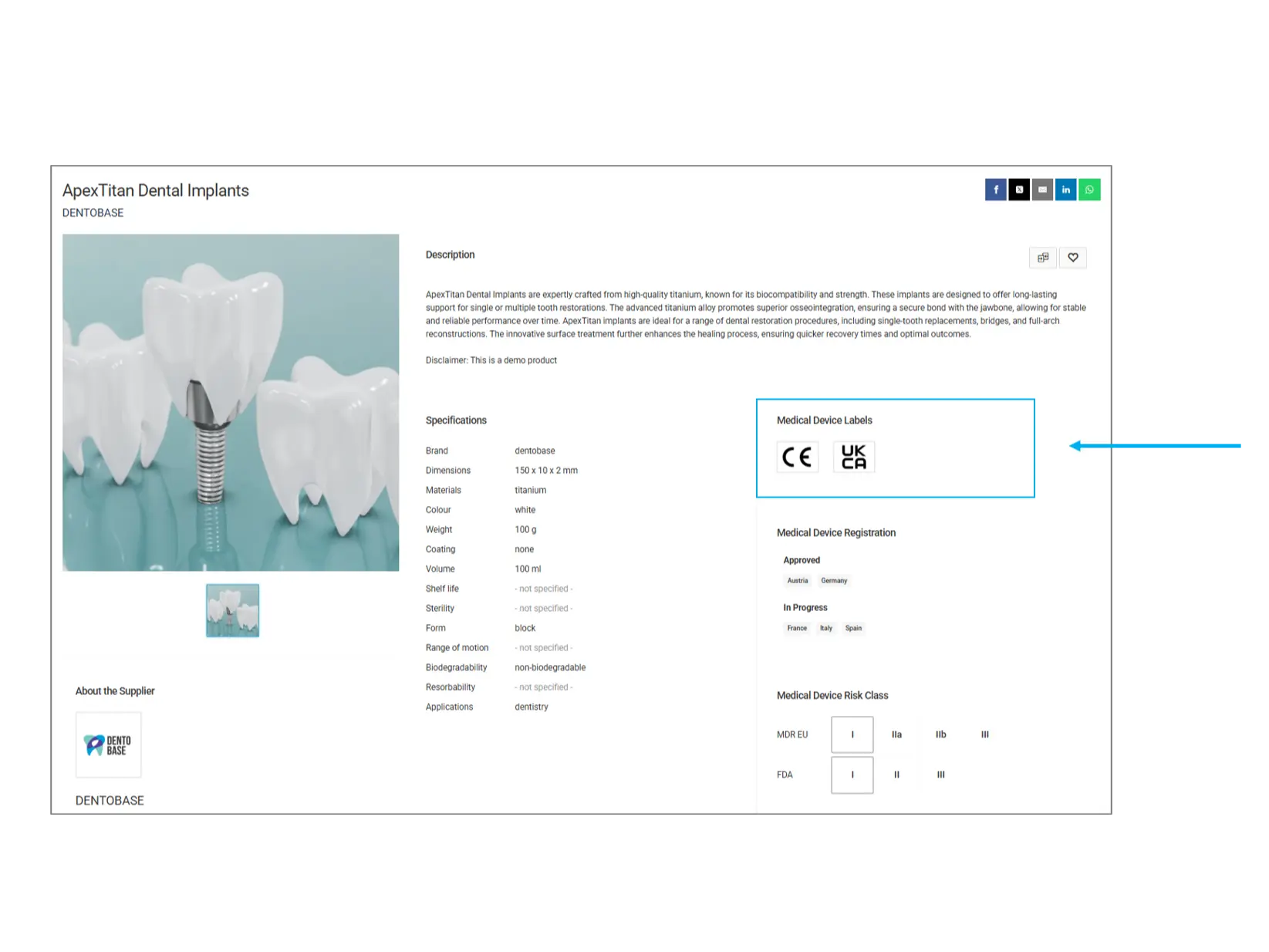
Medical Device Labels
Medical device labels indicate the regulatory approvals and standards a product complies with. These labels help ensure the product meets the necessary safety and quality guidelines for its intended use.
CE (Conformité Européenne): Indicates that the product meets EU safety, health, and environmental protection standards.
UKCA (United Kingdom Conformity Assessed): A certification for products sold in Great Britain, ensuring compliance with UK regulations.
Other Labels: Additional certification marks might be present, such as FDA, ISO, or other region-specific compliance marks.
What to check? Ensure that the product has the required certification(s) based on your country or regulatory requirements.
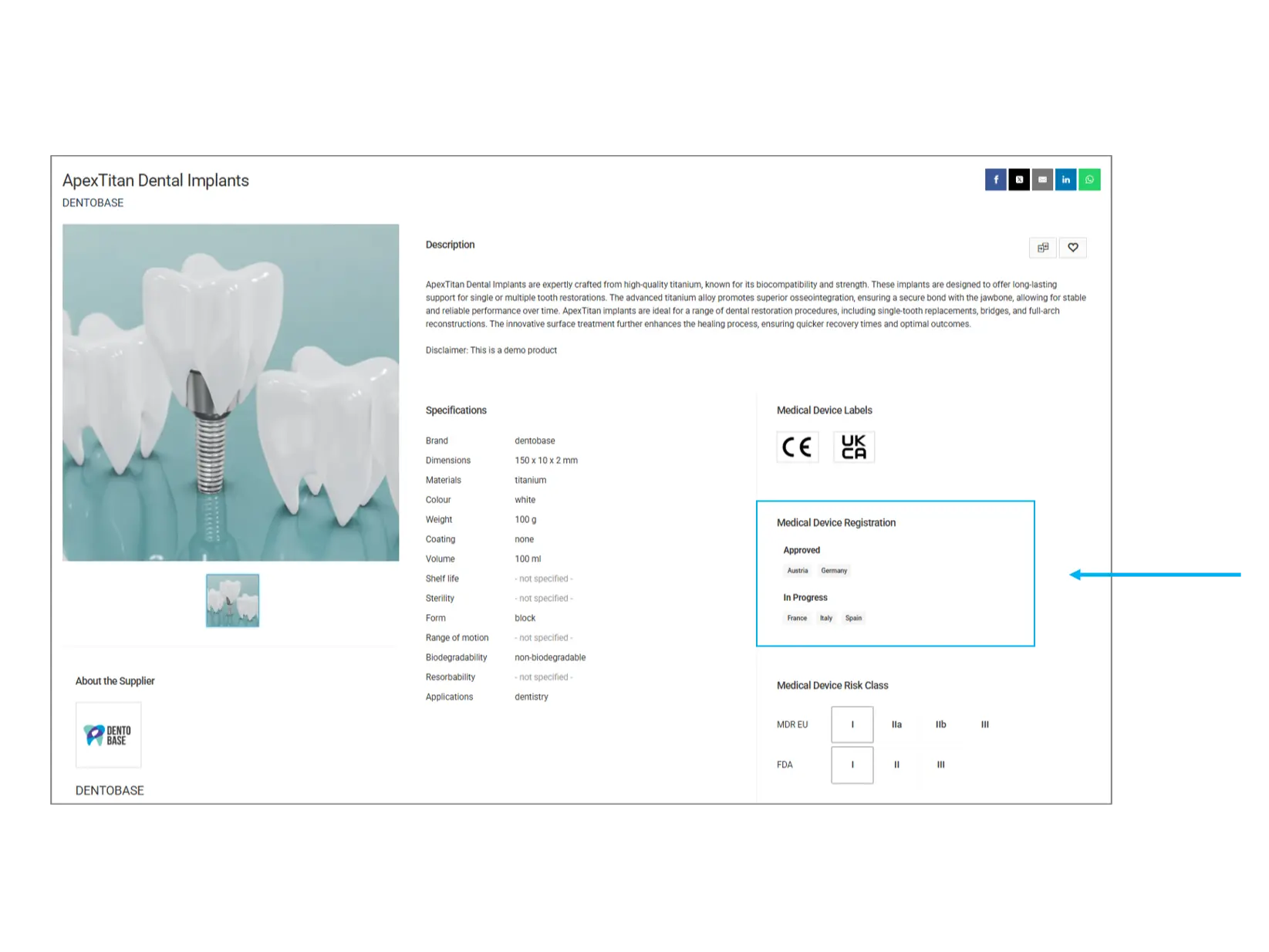
Medical Device Registration
This section provides details on the regulatory approval status of the product in different countries.
Approved Countries: The product has been officially registered and can be marketed in these regions.
In Progress: The product is undergoing regulatory evaluation and is not yet fully approved in the listed countries.
What to check? If you need a product for a specific market, confirm that it is approved in your country before purchasing.
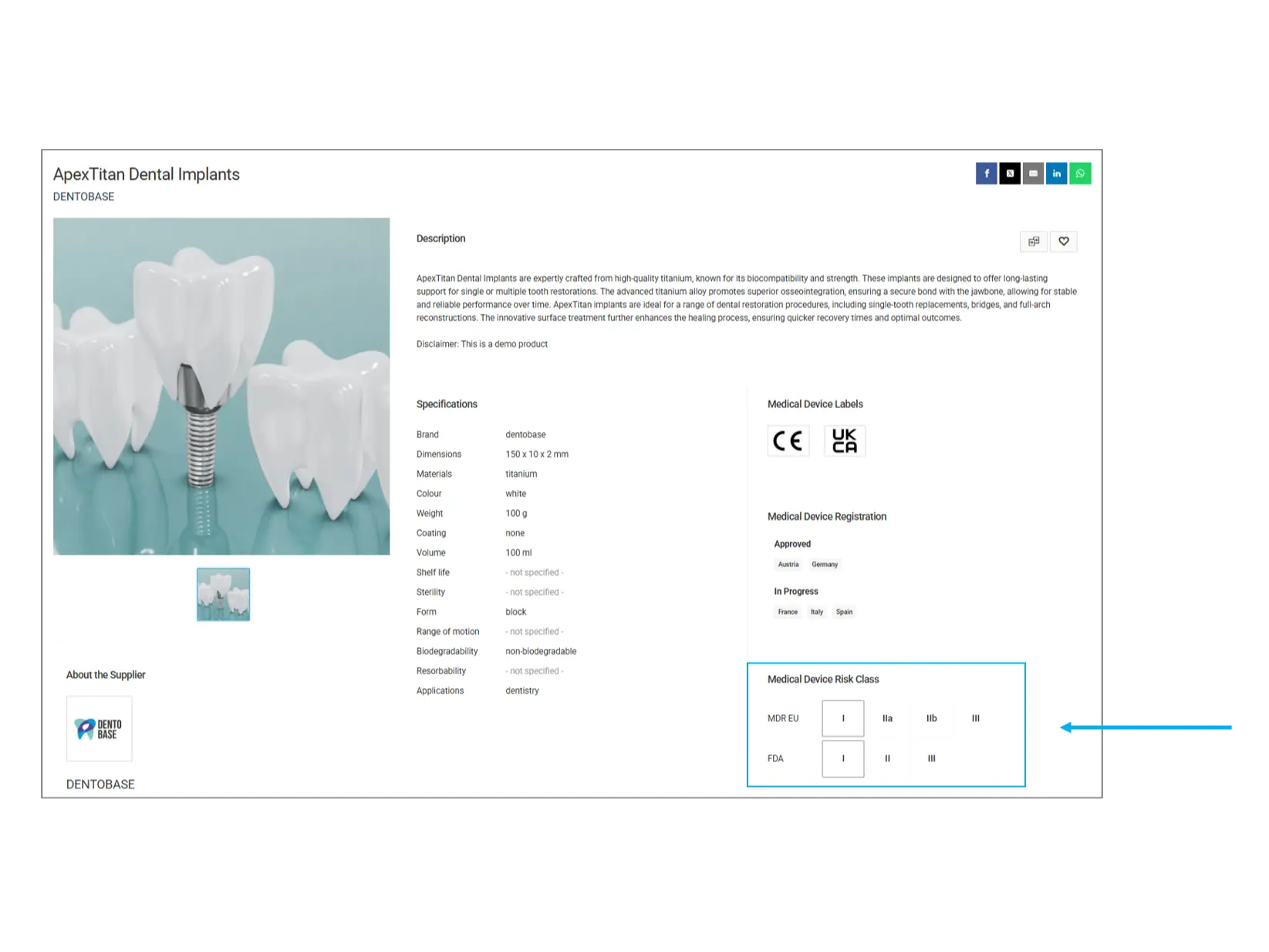
Medical Device Risk Class
Medical devices are categorized into different risk classes based on their potential impact on human health. The classification helps users understand the level of regulatory scrutiny and potential risks associated with the product.
MDR EU (Medical Device Regulation - European Union):
Class I: Low-risk devices (e.g., bandages, dental tools)
Class IIa: Medium-risk devices (e.g., hearing aids, diagnostic imaging)
Class IIb: Higher-risk devices (e.g., infusion pumps, ventilators)
Class III: High-risk devices (e.g., pacemakers, artificial heart valves)
FDA (U.S. Food and Drug Administration):
Class I: General controls (e.g., tongue depressors, surgical gloves)
Class II: Special controls (e.g., blood pressure cuffs, infusion pumps)
Class III: Premarket approval required (e.g., implantable defibrillators, prosthetic heart valves)
What to check? Higher-class devices undergo stricter regulations and testing. If your application requires a highly regulated product, ensure it meets the necessary classification.
Is this article helpful?